

What is J-DOIT3?
Introduction
Japan is rapidly on the way to the supper-aging society, and it is predicted that, by the year 2015, the number of elderly people will reach 33 million. Under these circumstances, to pursue life extension itself is not enough; we should aim at building a bright and vigorous society, in which all the people can stay healthy and active throughout their lives.
From this viewpoint, the Ministry of Health, Labour and Welfare has drawn up the Health Frontier Strategy, for prolongation of people's healthy lifespan. It was decided to carry out large-scale clinical trials, with the aim of improvement in incidence and mortality of lifestyle-related diseases, as well as reduction of patients requiring long-term nursing care.
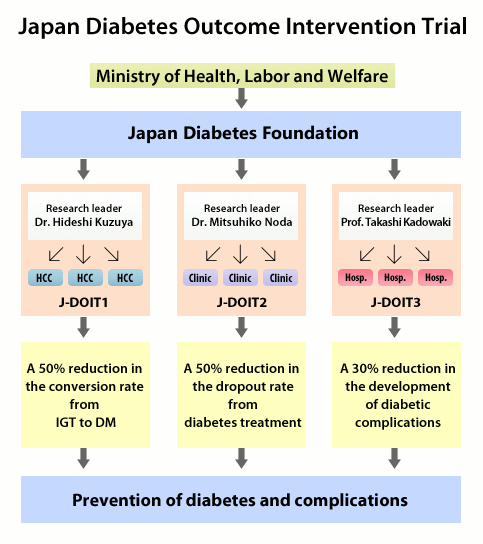
Among them, as for diabetes, Japan Diabetes Outcome Intervention Trial (J-DOIT)1, 2 and 3 were designed. J-DOIT1, or “Intervention study to prevent or delay type 2 diabetes mellitus,” is to identify how to suppress conversion from impaired glucose tolerance to diabetes, whereas J-DOIT2, or “The large trial to improve the medical care of the family doctors for type 2 diabetes patients and to decrease dropout rate of these patients” is to find how to reduce discontinuation of medical care for diabetes by primary care doctors, and J-DOIT3 is to elucidate how to prevent diabetic complications.
Background of J-DOIT3
In diabetes, insufficient insulin action induces abnormalities in glucose and lipid metabolism. If it lasts for a certain period, vascular complications common in diabetes appear, resulting in vision loss, renal failure and neuropathy, due to microvasclucar diseases, and myocardial infarction and stroke, due to macrovasclucar diseases.
Although some clinical trials have actually succeeded in suppressing microangiopathies, it was still unclear how diabetes should be treated in order for macroangiopathies to be prevented.
That is why J-DOIT3 was designed, targeting diabetic patients with hypertension and/or dyslipidemia who were at high risk of diabetic complications including macrovascular diseases.
Overview of J-DOIT3
The subjects of this trial are type 2 diabetes patients who also suffered from hypertension and/or dyslipidemia. The participants are to be randomly allocated into two groups: the conventional therapy group to undergo the current standardized treatment, and the intensive therapy group to undergo treatment for stricter control targets, with enhanced diet therapy, exercise theapy and medication. Both groups are to be followed up, to see any difference in diabetic complications, as follows.
| Targets | 2,542 subjects with type 2 diabetes (45-69 years old, HbA1c ≥ 6.9%) with hypertension and/or dyslipidemia. (Primary prevention 90%, secondary prevention 10%) |
|---|---|
| Primary endpoint | Occurrence of either of myocardial infarction, coronary bypass surgery, percutaneous transluminal coronary angioplasty, stroke, percutaneous transluminal cerebral angioplasty, carotid endarterectomy, carotid artery stenting, or death. |
| Secondary endpoints | (1) Occurrence of either of myocardial infarction, stroke, or death (2) Onset or progression of nephropathy (3) Occurrence of lower limb vascular events (amputation orrevascularization of lower limb) (4) Onset or progression of retinopathy |
| Study duration | Until the number of primary endpoint event reaches 250 (expected to be in March 2016). |
| Targets | Intensive Therapy Group (n=1,271) |
Conventional Therapy Group (n=1,271) |
|---|---|---|
| Blood glucose | HbA1c < 6.2% (pioglitazone-based therapy) |
HbA1c < 6.9% |
| Blood pressure | < 120/75mmHg (ARB/ACEI-based therapy) |
< 130/80mmHg |
| Lipids | LDL-C < 80mg/dL (*LDL-C < 70mg/dL) TG < 120mg/dL HDL ≥ 40mg/dL (Strong statin-basis) |
LDL-C < 120mg/dL (*LDL-C < 100mg/dL) TG < 150mg/dL HDL ≥ 40mg/dL * with the history of CHD |
Lifestyle modifications in the intensive therapy group are:
- Daily self-measurement of body weight.
- Diet therapy: regular nutritional counseling offered by registered dieticians, no between-meal eating, no late-evening snack, and cutting down of alcohol drinking.
- Exercise therapy, with monitoring of consumed calories and the number of steps with an accelerometer to be lent.
- Daily self-monitoring of blood glucose, with a blood glucose meter to be lent.
- Daily self-monitoring of blood pressure, with a sphygmomanometer to be lent.
- Cessation of smoking (in case of smokers).
The participants are to be asked to report the results of self-monitoring to the doctor in charge, and undergo treatment accordingly.

For more details, see ClinicalTrials.gov, registration page.(Clinicaltrials. gov Identifier : NCT00300976).
J-DOIT3 Outline
1. Outline of the clinical trial protocol
1.1 Objectives
As
an attempt to address one of many research tasks that remain highly unmet in a
large Japanese population and thus call for definitive solutions, the present
outcome trial represents a large-scale “strategic clinical study” with clearly
defined outcome goals, which has been designed to address one of the three
major tasks as part of the “Strategic Studies Aimed at the Prophylaxis of
Diabetes Mellitus” as defined in the Ministry of Health, Labor and
Welfare-designated scientific research project, “Research on Development of
Strategic Outcome Studies”, in a randomized controlled trial in type 2 diabetic
patients to verify the hypothesis that intensive multi-factorial therapy is
superior to conventional therapy in preventing the onset and progression of
vascular complications associated with diabetes, with the primary endpoint
being the occurrence of either of myocardial infarction, coronary bypass
surgery, percutaneous transluminal coronary angioplasty, stroke, percutaneous
transluminal cerebral angioplasty, carotid endarterectomy, carotid artery
stenting, or death.
1.2 Inclusion criteria
Subjects
were considered eligible for inclusion if they were 45 years old or older but
younger than 70 years old at study entry, had type 2 diabetes and met both “(1)
and (2)” or both “(1) and (3)” described below. Those who met all three “(1),
(2) and (3)” were also considered eligible for inclusion.
(1) Glycemic control
Those
with HbA1c 6.9% or greater despite treatment with any of the three regimens
given below.
j Diet and exercise
therapy alone
k Diet and exercise
therapy plus 1 oral anti-diabetic drug
l Diet and exercise therapy
plus aGI and 1 other oral
anti-anti-diabetic drug
(2)
Blood pressure control
Those
with the following casual blood pressure (BP) level as measured on an
outpatient basis
j Systolic BP ³ 140 mmHg or diastolic BP ³ 90 mmHg while not
on an antihypertensive agent
k Systolic BP ³ 130 mmHg or diastolic BP ³ 80 mmHg while on
1 or 2 ARB, ACEI or long-acting CCB
Those
receiving antihypertensive agents other than ARB, ACEI or long-acting CCB were
not eligible for study entry, with the exception of those who were receiving
these agents for other purposes than blood pressure lowering.
(3) Lipid control
Those
with the following fasting lipid levels while not on a lipid-lowering agent
j LDL-cholesterol, ³ 120 mg/dL (as estimated by using the Friedewald formula)
k Triglycerides, ³ 150 mg/dL
l HDL-cholesterol,
< 40 mg/dL
Subjects
receiving 1 lipid-lowering agent were judged eligible for study entry if they
met any of the above criteria. However, those on fibrates were to discontinue
that fibrate treatment at the start of the study when they were assigned to the
intensive therapy arm.
1.3 Exclusion criteria
1.
Those with poorly controlled hypertension despite pharmacological therapy
(systolic BP ³ 200 mmHg or
diastolic BP ³ 120 mmHg)
2.
Those on insulin therapy
3.
Those with non-diabetic renal disease
4.
Those in whom type 1 and other diabetes due to pathogenic mechanisms other than
those associated with type 2 diabetes is strongly suspected
5.
Those who tested anti-GAD antibody-positive
6.
Those with LDL-cholesterol ³ 200 mg/dL
7.
Those suspected of having secondary hypertension other than renal parenchymal hypertension
8.
Those suspected of having hereditary lipid disorder with a strong family
history of lipid metabolic disorder
9. Those
who were receiving antihypertensive agents other than ARB, ACEI, long-acting
CCB, except where they were receiving these agents for other purposes than
blood pressure lowering
10.
Those who were receiving 3 or more antihypertensive agents (i.e., ARB, ACEI,
and long-acting CCB), except where they were receiving these agents for other
purposes than blood pressure lowering
11.
Those with more serious retinopathy than proliferative retinopathy
12.
Renal failure (serum Cr: ³ 2.0 mg/dL in men;
³ 1.5 mg/dL in women)
13.
Those with a history of cardiac failure or those with cardiac failure
14.
Those who were pregnant or potentially pregnant
15.
Those who met any of the following criteria and who had BNP ³ 100 pg/mL
l
Myocardial infarction
l
Angina pectoris (or a history of disease)
l
History of coronary artery bypass graft (CABG)
l
History of percutaneous coronary angioplasty (PTCA)
l
Other cardiac disease
l
ECG findings of left ventricular hyperplasia
l
Abnormal ECG findings (excluding isolated
extrasystole or right bundle branch block [RBBB])
16.
Those judged by the attending physician to be ineligible for study entry
1.4 Investigational treatment
Subjects judged eligible for the study were randomly assigned to intensive multi-factorial therapy or conventional therapy. The management goals set for either therapy were described below.
The management goals for
intensive therapy are as follows. Weight reduction: BMI, £ 22; diet restriction: total energy intake to be rigorously controlled
(25 kcal/kg in those with BMI ³ 25; 27 kcal/kg in
those with BMI < 25) with lipid intake accounting for less than 25% of total
energy intake, cholesterol intake accounting for 300 mg/day or less, and salt
intake accounting for 6 g/day or less; alcohol and tobacco abstinence to be
rigorously adhered to; exercise therapy: 2 or more 15- to 30-minute walks on a
daily basis; glycemic control: HbA1c,
< 6.2%; blood pressure control: systolic BP, < 120 mmHg and diastolic BP,
< 75 mmHg; lipid control: LDL-C,
< 80 mg/dL*1, TG, < 120 mg/dL, HDL-C, ³ 40 mg/dL, except
in those with a history of coronary artery disease (CAD), where the goal is to
be set at LDL-C < 70 mg/dL*1.
The management goals for conventional
therapy are as follows. Weight reduction: BMI, £ 24; diet
restriction: appropriate diet therapy in accordance with the Japan Diabetes
Society Clinical Practice Guidelines; exercise therapy: appropriate exercise
therapy in accordance with the Japan Diabetes Society Clinical Practice
Guidelines; glycemic control: HbA1c,
< 6.9%; blood pressure control: systolic BP, < 130 mmHg and diastolic BP,
< 80 mmHg; lipid control: LDL-C,
< 120 mg/dL, TG, < 150 mg/dL, except in those with a history of CAD,
where the goal is to be set at LDL-C < 100 mg/dL.
*1 In those with TG ³ 150 mg/dL, the subjects are to be assessed in terms of non-HDL-C (=
total cholesterol – HDL-C) levels, with the management goal set as non-HDL-C
< 110 mg/dL, except in those with a history of CAD, where it is set as
non-HDL-C < 100 mg/dL.
1.5 Physical and laboratory examinations during planned patient visits
Physical
and clinical examinations during planned patient visits include:
Body
weight, blood pressure
control;
Blood
glucose, HbA1c (only stable HbA1c to be measured for adjustment against
reference material);
Total
cholesterol, LDL-cholesterol (non-HDL-cholesterol), HDL-cholesterol,
triglycerides;
Hematologic
test (white blood cell count, red blood cell count, hemoglobin, hematocrit,
platelet count), hepatic/renal function test (AST, ALT, g-GPT, LDH, BUN, serum Cr), serum electrolytes (Na, K, Cl), CPK; and
Urinary albumin, and urinary Cr (to be measured every 6 months).
1.6 Primary endpoint of the study
The
primary endpoint of the study is defined as the occurrence of either of myocardial
infarction, coronary bypass surgery, percutaneous transluminal coronary
angioplasty, stroke, carotid endarterectomy, percutaneous transluminal cerebral
angioplasty, carotid artery stenting, or death (irrespective of its causes).
1.7 Secondary endpoints of the study
The
secondary endpoints of the study include:
j Occurrence of either
of myocardial infarction, stroke or death
k Onset or
progression of nephropathy
l Lower limb
vascular events (amputation or revascularization of lower limb)
„ Onset or progression of
retinopathy
1.8 The trial duration and number of participating healthcare institutions
The
study is to remain open for patient accrual for a period of 2.75 years until
March 2009, with the subjects being followed up until the number of events for
the primary endpoint reaches 250 (expected duration from participation to March
2016).
1.9 Target patient accrual
The
target patient accrual is projected at 1,669 each for intensive and
conventional therapy, totaling 3,338 patients.
Note:
The actual number of subjects totaled 2,542 patients, or 1,271 patients each
for intensive and conventional therapy.
2. Investigational treatment
The
investigational treatment in this study is defined as “intensive
multi-factorial therapy” with “conventional therapy” serving as the
reference/control treatment, where each treatment is intended to achieve
lifestyle modification (weight reduction, diet restriction, exercise therapy
and smoking cessation) as well as glycemic, blood pressure and lipid control as
described below. In using pharmacologic agents for glycemic, blood pressure and
lipid control, it is essential that relevant package inserts be consulted to
ensure that they are not used in patients in whom their use is contraindicated.
2.1 Guidance on lifestyle modification
2.1.1 Weight reduction
All subjects in
both treatment arms are to be provided with patient diaries to record their own
body weight.
In the intensive therapy arm,
where the management goal for BMI is set for 22 or less, all subjects are
obliged to measure their body weight at a predetermined time once daily, record
and report on the measurement, and the physician in charge is to keep informed
of these measurements.
In the conventional therapy
arm, where the management goal for BMI is set for 24 or less, all subjects are
also obliged, as are those in the investigational treatment arm, to measure
their body weight, record and report on the measurement, and the physician in
charge is to keep informed of these measurements.
Once the management goal for
BMI (body weight) has been achieved in any patients in either arm, BMI is to be
maintained at that level. In those complicating diabetic nephropathy, the
management goal is to be achieved by drawing on the diet criteria for diabetic
nephropathy.
Between-meal and bedtime
snacks are to be prohibited as a rule, except where they are required to avoid
hypoglycemia associated with the use of oral anti-diabetic drugs or insulin
therapy.
2.1.2 Diet restriction
Total energy
intake is to be rigorously controlled in the subjects in the intensive therapy
arm. Specifically, in those with BMI 25 or greater at study entry, the
management goal for diet restriction is set for “25 kcal/1 kg of ideal body
weight (IBW)”, and in those with BMI less than 25, the goal for diet restriction
is set for “27 kcal/1 kg of ideal body weight (IBW)”.
In this regard, the subjects
in the intensive therapy arm are to be instructed in person that through diet restriction,
lipid is to account for less than 25% of total energy intake, with cholesterol
intake accounting for 300 mg/day or less and salt intake accounting for 6 g/day
or less. Additionally, they are to be instructed that between-meal and bedtime
snacks are to be prohibited, and alcohol abstinence (equivalent to 180 mL of
sake or less) is to be adhered to, and they are obliged to report on alcohol
intake at regular hospital visits.
Additionally, they are to be
given 30-minute or longer guidance on nutrition by the designated national
registered dietitians at the start of the study, 1, 3, 6 and 12 months after
the start of the study and every 6 months in the second year afterwards (a
separate manual on nutritional guidance is to be developed to ensure consistency
in their approach).
The subjects in
the conventional therapy arm are to be instructed about the total energy intake
to be adhered to in accordance with the Japan Diabetes Society Clinical
Practice Guidelines (hereafter Guidelines). Salt restriction is also to be
encouraged as required.
Subjects in both
arms are to be instructed on allowable diets using the “Food Exchange Lists -
Dietary Guidance for Persons with Diabetes (7th edition)” edited by
the Japan Diabetes Society. Again, those with diabetic nephropathy in both arms
are to be encouraged to restrict protein intake in accordance with the “A Guide
to the Management of Diabetes” edited by the Japan Diabetes Society.
2.1.3 Exercise therapy
All subjects in
both treatment arms are to be loaned an accelerometer at study entry.
The subjects in the intensive
therapy arm are to be instructed to walk 15 to 30 minutes twice or more
frequently per day. As a rule, the subjects are obliged to walk every day and
to report on the calorie consumed as well as the number of steps taken every
day. The physicians or nurses in charge are to keep informed of their activity
status so that it can be borne in mind when giving exercise instructions.
The subjects in the
conventional therapy arm are to be instructed in accordance with the
Guidelines.
2.1.4 Smoking cessation
All subjects in
the intensive therapy arm are to be instructed to persevere in their smoking
cessation practice and to report on the number of cigarettes smoked at their
regular hospital visits. Those who find smoking cessation difficult are to be
encouraged to use smoking-cessation aids.
2.1.5 Implementation of the core program
To ensure that the
goals for lifestyle modification (weight reduction, dietary restriction,
exercise and smoking cessation) are met n the intensive therapy arm, a core
program intended to improve lifestyle habits is to be implemented to ensure
lifestyle factors are improved in the subjects. A post-core program will also
be implemented after completion of the core program to ensure lifestyle factors
are further improved in the subjects. Details of the core program are given
elsewhere.
2.2 Glycemic control
2.2.1 Intensive therapy
In the intensive therapy arm, the goal for glycemic control is defined as HbA1c < 6.2% and the subjects are to make regular hospital visits for glycemic control and monitoring (see Note 1). If any of the subjects receiving treatment on an outpatient basis does not achieve the management goals, they will be immediately hospitalized to ensure better glycemic control in these subjects.
Furthermore, all subjects in the intensive therapy arm are to be loaned a blood glucose meter for self-monitoring of blood glucose (SMBG), and the physicians in charge are to keep informed of the measured results in these subjects.
All subjects are to be treated first with predetermined diet and exercise therapy, and if they fail to achieve HbA1c < 6.2% in the first 3 months, they will be given anti-diabetic drugs in the following steps (those already on anti-diabetic drugs at study entry are to continue with the medication and be treated with lifestyle modification to achieve their respective management goals). Anti-diabetic drugs to be used include thiazolidinedione derivatives, biguanides, sulfonylureas, rapid-onset insulin secretagogues (glinides), a-glucosidase inhibitors, DPP-4 inhibitors, SGLT2 inhibitors, GLP-1 receptor antagonists and insulin. Of these, sulfonylureas and glinides are both designated as “insulin secretagogues” and DPP-4 inhibitors and GLP-1 receptor agonists are designated as “incretin-related drugs.”
The treatment specified in each of the steps is to be continued if the treatment leads to HbA1c < 6.2% or a ³ 1% decrease in HbA1c within 6 months for the subjects. If, however, the goal of HbA1c < 6.2% cannot be achieved with treatment and if treatment is associated with a less than 1% decrease in HbA1c, the treatment is to be intensified to the next step (refer to Notes 1 and 2 if only one of the goals is achieved). In any of the steps described, the dosage may be increased, decreased, discontinued or changed (see Note 2), and an a-glucosidase inhibitor, DPP-4 inhibitor, or SGLT2 inhibitor may be added at the discretion of the physician in charge. For instructions on the addition of a biguanide or GLP-1 receptor agonist, see Note 3 and, for directions for use of incretin-related drugs, see Note 4.
The thiazolidinedione derivative pioglitazone is to be given up to 30 mg/day as a standard maximal dose but the dose can be further increased to 45 mg/day if the physician in charge finds it necessary. However, utmost care is to be taken in increasing the dose from 30 mg to 45 mg/day.
Again, if the physician in charge finds it necessary, temporary or continued insulin therapy can be given (this constitutes step 3). In this case, however, a thiazolidinedione derivative is to be used in combination as long as that is possible. Whether or not to use a thiazolidinedione in combination is to be decided at the discretion of the physician in charge.
In any of the steps described below, diet and exercise therapy is to be continued.
Step 0:
If the goal of obtaining an HbA1c < 6.2% is achieved with diet and exercise therapy plus or minus an a-glucosidase inhibitor, DPP-4 inhibitor and/or SGLT2 inhibitor within the first 3 months, that particular treatment is to be continued (step 0).
Step 1:
The subjects are to be treated with a drug from among those listed below according to their BMI values at study entry. However, thiazolidinedione derivatives associated with a certain level of evidence that supports their use are to be used whenever possible. If any of the subjects are already receiving the drug at study entry, the treatment is intensified to step 2.
j A thiazolidinedione derivative (or possibly a biguanide or GLP-1 receptor agonist) in those with BMI ³ 25
k A thiazolidinedione derivative (or possibly a biguanide or GLP-1 receptor agonist) or an insulin secretagogue in those with 22 £ BMI < 25
l An insulin secretagogue or thiazolidinedione derivative (or possibly a biguanide or GLP-1 receptor agonist) in those with BMI < 22
Step 2:
Depending on the drug used at study entry or in step 1, an appropriate choice (j or k) is to be made.
j Add an insulin secretagogue if a thiazolidinedione derivative (biguanide or GLP-1 receptor agonist) is already being used
k Add a thiazolidinedione (or possibly a biguanide or GLP-1 receptor agonist) if an insulin secretagogue is already being used
Step 3:
Insulin therapy is to be initiated. The insulin regimen may be chosen from among the following, and also changed during the course of treatment, in accordance with the patient’s symptoms:
j Intermediate- or long-acting insulin injection once daily before bedtime
k Intermediate- or mixed insulin injection twice daily (before breakfast and dinner)
l Rapid- or ultra-rapid-onset injection insulin three times daily (before meals)
m Rapid- or ultra-rapid-onset insulin injection three times daily (before meals) plus intermediate- or long-acting insulin injection once daily before bedtime
All subjects are to be given a thiazolidinedione derivative whenever possible even after they have moved on to insulin therapy. The physician in charge is to be responsible for any decision made on the use of an anti-diabetic drug other than a thiazolidinedione derivative in combination with insulin therapy.
Note 1: The frequency of hospital visits will be at least once a month until March 2010 and, thereafter, as set forth below.
j As a rule, at least once a month in those patients receiving insulin therapy and those patients with unstable glycemic control.
k Maximum of 3 months apart in those patients who have almost achieved management goals for glycemic control and other factors and whose condition is stable (except patients receiving insulin therapy).
Note 2: The physician in charge may at any time replace one insulin secretagogue with another at his/her discretion as required, i.e., replacing a sulfonylurea with a glinide or vice versa. Additionally, the physician in charge may at any time switch among a thiazolidinedione, biguanide, or GLP-1 receptor agonist or use a combination thereof, at his discretion. Also, the physician in charge may at any time replace one drug with another within the same drug class.
Note 3: When a biguanide or GLP-1 receptor agonist has been added, the treatment is intensified to the next step, except in the following cases:
l When a biguanide or GLP-1 receptor agonist was given in subjects with HbA1c < 6.2%
l When a biguanide or GLP-1 receptor agonist was given in subjects receiving a thiazolidinedione derivative
When adequate glycemic control has not been achieved with a biguanide combined with a GLP-1 receptor agonist and/or an insulin secretagogue, a thiazolidinedione derivative is to be given whenever possible. In which case, where a thiazolidinedione derivative was given to subjects already receiving a biguanide or a GLP-1 receptor, the duration of therapy for step 2 is to be 6 months from the time when the thiazolidinedione derivative was started.
Note 4: When an incretin-related drug is used in combination with an SU, etc., the drugs should be administered in accordance with the respective Package Insert for each drug while paying careful attention to the occurrence of hypoglycemia. Moreover, incretin-related drugs should be administered in accordance with the Recommendations on Appropriate Use of Incretin-Related Drugs.
Note 5: If an SGLT2 inhibitor is administered, the drug should be used in accordance with the Package Insert for the drug while paying careful attention to the onset of dehydration or other symptoms. If an SGLT2 inhibitor is used in combination with insulin or an SU, etc., the drugs should be administered in accordance with the respective Package Inserts for each drug while paying careful attention to the onset of hypoglycemia. Moreover, SGLT2 inhibitors should be administered in accordance with the Recommendations on Appropriate Use of SGLT2 Inhibitors.
Dose reduction and discontinuation of
anti-diabetic drugs
The physician in charge is to carefully watch for episodes of hypoglycemia in all subjects receiving anti-diabetic drugs, and to adequately educate the subjects on how to deal with these episodes when they appear. In the subjects with frequent hypoglycemia, every effort is to be made to immediately optimize drug therapy by having them consult the physician in charge as often as possible or by hospitalizing them. In optimizing drug therapy, the decision as to which drug needs to be discontinued or reduced is to be left up to the physician in charge. However, even when the physician in charge makes such a decision, he/she is to avoid discontinuing the thiazolidinedione derivative already being used whenever possible. When fluid retention occurs likely due to the use of a thiazolidinedione derivative, the physician in charge is to deal with it by giving a diuretic or by reducing or discontinuing the thiazolidinedione derivative at his discretion.
Note 1: If the goal of HbA1c < 6.2% has been achieved with drug therapy, that particular treatment is to be continued. However, the decision is left up to the physician in charge on whether or not to increase, reduce or discontinue a particular drug, replace a sulfonylurea with a glinide or vice versa, switch treatment among a thiazolidinedione derivative, biguanide, or GLP-1 receptor agonist, replace a drug with another within the same drug class, or add an a-glucosidase inhibitor, DPP-4 inhibitor, SGLT2 inhibitor, biguanide, or GLP-1 receptor agonist. When HbA1c becomes 6.2% or greater in a particular subject, every effort is to be made to ensure that HbA1c is maintained below 6.2% by implementing more intensive therapy (more rigorous lifestyle modification, dose increases or replacements). In the event that HbA1c becomes 6.2% or greater on 3 consecutive measurements despite this intensive therapy in a particular subject, it is to be deemed difficult to achieve the predetermined management goals in this patient, the treatment is intensified to the next step depending on the management goals achieved in this patient as follows:
Diet/exercise therapy (+a-glucosidase inhibitor, DPP-4 inhibitor and/or SGLT2 inhibitor) (considered as step 0) " step 1
1 anti-diabetic drug (+a-glucosidase inhibitor, DPP-4 inhibitor and/or SGLT2 inhibitor) (considered as step 1) " step 2
2 anti-diabetic drugs (+a-glucosidase inhibitor, DPP-4 inhibitor and/or SGLT2 inhibitor) (considered as step 2) " step 3
Note 2: If HbA1c has decreased by more than 1% in a particular patient in the first 6 months, the same treatment in the treatment step is to be continued to ensure that HbA1c is maintained below 6.2% or decreases by more than 1% in the next 6 months.
Note 3: When a particular patient was started on insulin therapy, this is to be construed as constituting step 3. If that patient stops requiring insulin therapy at a later date, the patient is to be construed as being stepped down to that which is consistent with the drug therapy he/she happens to be receiving at that time.
2.2.2 Conventional therapy
The goal for
glycemic control in the conventional therapy arm is defined as HbA1c < 6.9%,
and the physician in charge is to administer appropriate therapy in accordance
with the Guidelines.
2.3 Blood pressure control
All the subjects
in both treatment arms are to be loaned a blood pressure manometer at study
entry.
2.3.1 Intensive therapy
The goal for blood
pressure control in the intensive therapy arm is defined as “systolic blood
pressure < 120 mmHg and diastolic blood pressure < 75 mmHg” as measured
on an outpatient basis.
All subjects in the intensive
therapy arm are obliged to measure their blood pressure control by using the
loaned blood pressure manometer, record and report on the measurement, and the
physician in charge is to keep informed of these measurements as a basis for
decisions regarding antihypertensive drug dose escalation or replacement. All
subjects are to be treated first with predetermined diet and exercise therapy
(those already on antihypertensive drugs at study entry are to continue with
the medication to achieve their management goal), and if they fail to achieve
the blood pressure goal in the first 3 months, they will be given
antihypertensive drugs in the following steps to ensure that the blood pressure
goal is achieved in 3 to 6 months. However, in those with systolic blood
pressure 160 mmHg or greater or diastolic blood pressure 100 mmHg or greater,
diet and exercise therapy, where blood pressure control is to be only
monitored, is to be skipped, and antihypertensive therapy is to be immediately
initiated by following the steps shown below.
In any of the steps described below, diet and exercise therapy is to be continued.
Step 1:
Give an
angiotensin receptor blocker (ARB) or angiotensin-converting enzyme inhibitor
(ACEI) at its standard dose with the dose being escalated to its maximal dose as
required until the blood pressure goal is achieved in a particular subject. If
the goal cannot be achieved even with the maximal dose of the ARB or ACEI, the
treatment is intensified to step 2.
Step 2:
Add a long-acting
calcium channel blocker (CCB) to the antihypertensive drug used, with the dose
being escalated as required. Those already receiving a long-acting CCB, the
treatment is intensified to step 3.
Step 3:
Add on a diuretic,
b-blocker or a-blocker, one agent at a time as required, in this order.
Drug dose
reduction or discontinuation is to be decided at the discretion of the physician
in charge, where the decision as to which drug is to be discontinued or by how
much the dose needs to be reduced is to be left up to the physician in charge.
The physician in charge may at any time replace an ARB with an ACEI or the
other way around, or replace one drug with another within the same drug class
at his/her discretion.
In those receiving 1
long-acting CCB at study entry and in whom the blood pressure goal cannot be
achieved within the first 3 months, the long-acting CCB is to be switched to an
ARB or ACEI (step 1). Again, if the goal for blood pressure control could not
be achieved in these subjects even when an ARB or ACEI was added to the
long-acting CCB with its dose escalated to its maximal dose, they are to be
moved up to step 3. In this regard, the physician in charge may decide in which
step a particular subject is to be treated.
Blood pressure
control after achievement of the goal for blood pressure control
If desired blood pressure control is maintained with drug therapy in a particular patient beyond the period specified above, that particular treatment is to be continued, irrespective of which step the patient happens to be in (although the physician in charge may reduce the drug dose or discontinue the drug at his/her discretion).
If blood pressure control begins to exceed the blood pressure goal in this patient at a later date, the patient is to be treated as shown below.
If the patient is in step 1 or 2, the drug therapy designated in that treatment step is to be used with the drug dose being increased or another drug added or with enhanced diet and exercise therapy, where blood pressure control in excess of the blood pressure goal as confirmed on 3 consecutive measurements is to be construed as an indication for the next step up.
If the patient is in step 3, the patient is to be treated to ensure that he/she achieves the goal for blood pressure control, if possible, by increasing the dose of the drug he/she is receiving, adding another drug or enhancing the diet and exercise therapy being given.
Note: If a particular patient called for no blood pressure control at study entry and already achieved the blood pressure goal, he/she is to be treated in accordance with the treatment step in which he/she is being treated. However, if the patient called for no blood pressure control at study entry but has not achieved the blood pressure goal, he/she is to be treated consistently with the treatment step in which he/she is being treated and observed for 3 months. If the patient has achieved the blood pressure goal at the end of the observation period, the same treatment is to be continued; however, once his/her blood pressure begins to exceed the blood pressure goal, he/she is to be treated as shown below:
Those on no antihypertensive drugs are to be moved to step 1 to receive a standard dose of an angiotensin receptor blocker (ARB) or angiotensin-converting enzyme inhibitor (ACEI).
Those on an ARB or ACEI alone are to be moved to step 1 and treated with the dose being escalated as required to its maximal dose (Those on 2 ARB/ACEI or more are to be treated with the dose of one of these agents being escalated to its maximal dose).
Those on a long-acting CCB and ARB or ACEI are to be treated with the dose of the ARB or ACEI being escalated to its maximal dose, and to be moved up to step 3 if the treatment failed.
Those on a long acting CCB alone are to be switched to an ARB or ACEI and moved up to step 1. Alternatively, they are to be given a standard dose of an ARB or ACEI, with the dose being escalated as required to its maximal dose, and then to be moved up to step 3 if the treatment failed. The physician in charge may choose between these two options at his/her discretion.
2.3.2 Conventional therapy
The goal for blood
pressure control in the conventional therapy arm was defined as “systolic blood
pressure < 130 mmHg and diastolic blood pressure < 80 mmHg” as measured
on an outpatient basis. The physicians in charge are to choose appropriate
therapy for the subjects in the conventional therapy arm in accordance with the
Guidelines.
2.4 Lipid control
2.4.1 Intensive therapy
The goal for lipid
control in the intensive therapy arm is defined as “LDL-cholesterol (LDL-C)
< 80 mg/dL and triglycerides (TG) < 120 mg/dL and HDL-cholesterol (HDL-C)
³ 40 mg/dL”. However, for
those with a history of coronary artery disease (CAD), the goal for LDL-C
control is defined as < 70 mg/dL. In those with TG 150 mg/dL or greater,
lipid control is to be evaluated in terms of non-HDL-C (= total cholesterol –
HDL-C) values, instead of LDL-C values. In which case, the goal for non-HDL-C
control is defined as the goal for LDL-C control plus 30 mg/dL. In other words,
the goal for non-HDL-C control is defined as < 110 mg/dL in those with no
history of CAD and 100 mg/dL in those with a history of CAD.
All subjects are to be
treated first with predetermined diet and exercise therapy (those already on
lipid-lowering drugs at study entry are to continue with the medication, except
for fibrates that need to be discontinued, to achieve their management goal).
If they fail to achieve the lipid control goal in the first 3 months, they will
be given lipid-lowering drugs in the following steps.
In any of the steps described below, diet and exercise therapy is to be continued.
Step 1:
Give a standard
dose of atorvastatin, pitavastatin or rosuvastatin to all subjects who have not
achieved the goal for LDL-C (non-HDL-C) control or that for TG control.
Step 2:
The drug given in
step 1 is to be used with the dose being escalated as required to the upper
limit of the approved dosage.
Step 3:
Add on an
anion-exchange resin and/or omega-3 fatty acids in any order.
Omega-3 fatty
acids are to be given if the goal for TG control (< 120 mg/dL) has not been
achieved after the goal for LDL-C (non-HDL-C) goal has been achieved in step 1
or 2 or if the goal for TG control has not been achieved in step 3.
In all treatment steps,
attention is to be focused on improving hypo-HDL-cholesterolemia with exercise,
smoking cessation or a thiazolidinedione derivative when it occurs.
Drug dose
reduction or discontinuation is to be decided at the discretion of the physician
in charge, where the decision as to which drug to discontinue or by how much
the dose needs to be reduced is to be left up to the physician in charge. The
physician in charge may at any time replace atorvastatin with pitavastatin or
rosuvastatin, replace pitavastatin with atorvastatin or rosuvastatin, or replace
rosuvastatin with atorvastatin or pitavastatin at his/her discretion.
Lipid control
after achievement of the goal
If desired lipid control is maintained with drug therapy in a particular patient beyond the period specified above, that particular treatment is to be continued (although the physician in charge may reduce the drug dose or discontinue the drug at his/her discretion).
If lipid control levels begin to exceed their respective goals in this patient at a later date, the patient is to be treated as shown below.
If the patient is in step 1 or 2, the drug therapy designated in that treatment step is to be used with the drug dose being increased or another drug (omega-3 fatty acids) added or with enhanced diet and exercise therapy, where lipid control (LDL-C [non-HDL-C] and TG) levels in excess of their respective goals as confirmed on 3 consecutive measurements is to be construed as an indication for the next step up.
If the patient is in step 3, attention is to be focused on ensuring that he/she achieves the goal for lipid control, if possible, by increasing the dose of the drug he/she is receiving or enhancing the diet and exercise therapy being given.
Note: In the subjects who were receiving fibrates at study entry and were then assigned to the intensive therapy arm, all fibrates are to be discontinued. If a particular patient called for no lipid control at study entry and already achieved the lipid control goal, he/she is to be treated in accordance with the treatment step in which he/she is being treated. However, if the patient called for no lipid control at study entry but has not achieved the goal for lipid control, he/she is to be treated consistently with the treatment step in which he/she is being treated and observed for 3 months. If the patient has achieved the goal for lipid control at the end of the observation period, the same treatment is to be continued; however, once his/her lipid levels begin to exceed the goal for lipid control, all medications for dyslipidemia are to be discontinued and atorvastatin, pitavastatin or rosuvastin is to be started in the patient. Drug dose is to be determined at the discretion of the physician in charge, and the patient is to be treated in step 1 or 2 depending on the dose, with the subsequent treatment being organized in accordance with the treatment steps for lipid control.
2.4.2 Conventional therapy
The goal for lipid
control in the conventional therapy arm is defined as “LDL-cholesterol (LDL-C)
< 120 mg/dL and triglycerides (TG) < 150 mg/dL”. However, for those with
a history of coronary artery disease (CAD), the goal for LDL-C control is
defined as < 100 mg/dL. The physicians in charge are to choose appropriate
therapy for the subjects in the conventional therapy arm in accordance with the
Guidelines.
2.5 Other treatments
In both the
intensive therapy and conventional therapy arms, all subjects with a history of
cardiovascular disease are to be given anti-platelet and anti-coagulant therapy
such as low-dose aspirin (enteric tablets) in accordance with the Guidelines.
All subjects who are already receiving such therapy are to be given the same
treatment in both the intensive therapy and conventional therapy arms.
The physician in
charge may at his/her discretion use any drugs other than those defined in 8.2,
8.3 and 8.4 for glycemic, blood pressure and lipid control.
3. Observation/evaluation schedule
Measurement/evaluation
of blood pressure control, glucose level, HbA1c*4, total cholesterol, LDL-C*5
(non-HDL-C*6), HDL-C, TG, gematologic examinations*7, hepatic/renal function
test*8, CPK, serum electrolytes (Na, K*9, Cl) are scheduled at the time of informed
consent*2 (from the time of consent obtained until before interim study
registration), at final study registration, and at reglular hospital visits *3.
Hematologic examinations scheduled in fasting states at the time of informed
consent, at official study entry and every 12 months.
Measurement of body height, waist
circumference, and parameters to be examined centrally*13 are
scheduled at final study registration and every 12 months.
Measurement of body weight is
scheduled at final study registration and every 12 months.
Measurement of urinary
albumin*10, urinary creatinine (to be measured in the same sample)
is scheduled at final study registration, at regular hospital visits *11, and every
6 months.
Measurement of brain
natriuretic peptide (BNP) is scheduled at informed consent*12.
Chest X ray, ECG, fundus
examination are scheduled to be performed at informed consent and every 12
months.
*1 Hematologic
examinations scheduled at the time of informed consent (from the time of
consent obtained until before interim study registration), at official study
entry and every 12 months are to be performed in fasting states.
*2 Examinations
scheduled at the time of informed consent are to be performed from the date
consent obtained until before interim study registration.
Items
to be examined during this time include: blood pressure control, total
cholesterol, LDL-cholesterol (non-HDL-cholesterol), HDL-cholesterol,
triglycerides
Chest x ray, echocardiogram
(ECG), fundus examination (by an ophthalmologist)
(Available data on these
examinations that is up to 6 months old may alternatively be used)
Hepatic/renal function test
(serum creatinine), BNP (only in those suspected of meeting the relevant
exclusion criteria)
*3 These are to be
examined on a regular basis during the entire study period in conjunction with
the hospital visits planned for each subject.
*4 Only stable HbA1c
is to be measured and adjusted against reference material.
*5 To be estimated
by using the Friedewald formula (LDL-C = total cholesterol – HDL-C – TG ´ 1/5), except in those with TG levels ³ 400 mg/dL.
*6 To be estimated
by using the formula: non-HDL-C = total cholesterol – HDL-C
*7 This includes
white blood cell count, red blood cell count, hemoglobin, hematocrit, and
platelet count.
*8 This includes
AST, ALT, g-GPT, LDH, BUN,
and serum creatinine.
*9 Those in whom the
potassium level ranged between 5.6 and 5.9 mEq/L are to be re-examined. Those
in whom the potassium level exceeded 6.0 mEq/L are to be re-examined and
treated appropriately.
*10 Urinary albumin
is to be evaluated for amount of its excretion per 1 g of urinary creatinine,
and to be evaluated in morning urine samples at final study registration and
every 12 months.
*11 Those who had an
episode of nephropathy are to be re-examined within 3 months of its occurrence
using a morning urine sample.
*12 To be performed
only in those suspected of meeting the relevant exclusion criteria (including
myocardial infarction, angina or a history of angina, a history of coronary
bypass surgery, a history of percutaneous coronary angioplasty, other cardiac
disease, ECG findings of left ventricular hyperplasia, and abnormal ECG
findings)
*13 Items to be
examined and the timeline for these examinations are described in section 9.6.
Examination dates
l
Examinations scheduled
for implementation at patient hospital visits every 6 months and every 12
months (except chest X-ray, ECG and fundus exams) are to be performed between 1
month before the predetermined date and 1 month after that date.
l
Available chest x ray, ECG and fundus exams that are
up to 6 months old may alternatively be used. These examinations scheduled
every 12 months are to be performed between 2 months before the predetermined
date and 2 months after that date.
l
Final study registration is to be completed one day
after but within 3 months of the date of informed consent.
Questionnaire-based
surveys are to be conducted in subjects prior to final study registration as
well as after 1 and 3 years and at study completion. Subjects who discontinue
treatment will be surveyed at the time of discontinuation. In addition, a
cognitive function test will be performed at the end of the study. The details
of these surveys to be conducted are described in a separate document.
If an assessment
of risk factors or frequency of events, other complications, and adverse drug
reactions is required, additional parameters will be measured at the central
laboratory using residual samples or clinical information will be collected
using questionnaires or other tools.
4. Efficacy evaluation
4.1 Primary endpoint
The primary
endpoint of the study is defined as the “occurrence of either of myocardial
infarction, coronary bypass surgery, percutaneous transluminal coronary
angioplasty, stroke, carotid endarterectomy, percutaneous transluminal cerebral
angioplasty, carotid artery stenting, or death (irrespective of its causes)”.
4.2 Secondary endpoints
The secondary endpoints
of the study are defined as the following four events.
1. Occurrence of either
of myocardial infarction, stroke, or death
2. Onset or
progression of nephropathy
3. Occurrence of
lower limb vascular events (amputation or revascularization of lower limb)
4. Onset or
progression of retinopathy
4.3 Definition of the endpoints used
A) Myocardial
infarction
Myocardial
infarction is defined as the presence of typical symptoms (e.g., lasting severe
chest pain), echocardiographic changes, abnormal laboratory findings (e.g.,
elevation of the cardiac enzyme troponin T), or findings of clinical imaging
(e.g., coronary angiography, cardiac scintigraphy, multi-detector row computed
tomography) which were diagnosed as such by a physician.
B) Stroke
Stroke
is defined as the presence of a newly onset focal symptom lasting more than 24
hours which was diagnosed as such by a physician (preferably with the culprit
lesion identified/confirmed on CT or MRI/MRA).
C) Onset or
progression of nephropathy
Based
on urinary albumin (excretion per 1 g of urinary creatinine) classified into
Normoalbuminurea,
urinary albumin < 30 mg/g·Cr;
Microalbuminurea,
urinary albumin ³ 30 mg/g·Cr, < 300 mg/g·Cr; and
Macroalbuminurea,
urinary albumin ³ 300 mg/g·Cr,
When
either of the following occurs, that time point is to be construed as the
occurrence of an event.
a. Progression from
normoalbuminurea to microalbuminurea or from normoalbuminurea to
macroalbuminurea
b. Progression from
microalbuminurea to macroalbuminurea
c. Serum creatinine
elevated 2-fold or more compared to that at study entry
d. End-stage renal
failure (permanent dialysis initiated or renal transplant performed)
For
a. and b., if both the measured urinary albumin value and the value obtained
from a retest (using a morning urine sample) are more than 30% higher than that
at study entry, the occurrence is considered an event. For c, retesting will be
performed within 3 months.
In
a., b., or c., the event is to be evaluated in their first occurrence.
Additionally,
affected subjects are to be evaluated for annual decreases in glomerular
filtration rate as calculated from urinary albumin excretion rate and its
conversion formulas.
D) Onset or
progression of retinopathy
When
either of the following occurs unilaterally or bilaterally, that time point is
to be construed as the occurrence of an event.
a. Progression from
absence of retinopathy to non-proliferative retinopathy or proliferative
retinopathy
b. Progression from
non-proliferative to proliferative retinopathy
c. Loss of vision
likely due to retinopathy
4.4 Assessment of the endpoints
A separate
Endpoint Assessment Committee is to be put in place for macrovascular
complications, nephropathy, and retinopathy to verify each of the events that
constitute the primary and secondary endpoints of the study.
5. Safety evaluation
5.1 Technical terminology
In the present
study, “adverse reactions” and “side effects” are defined as followed.
l
Adverse reactions
Adverse
reactions are defined as any untoward (or unintended) symptoms and signs
(including abnormal laboratory findings) that may or may not be causally
related to the investigational treatment. In other words, all untoward symptoms
or signs that occurred in the study subjects, including those whose causal
relationship to the investigational treatment can be ruled out, are to be
recorded as “adverse reactions”.
In this
trial, all untoward laboratory findings that occurred after the start of the
study are to be handled as adverse reactions, in addition to all symptoms and
signs that newly occurred or worsened after the start of the study.
l
Side effects
Of
the untoward (or unintended) symptoms and signs (including abnormal laboratory
findings), those whose causal relationship to the investigational treatment
cannot be ruled out are defined as side effects. In other words, of the “adverse
reactions” defined above, those whose causal relationship to the
investigational treatment cannot be entirely ruled out are to be handled as
side effects.
5.2 Collection of data on adverse reactions
During the course
of the study, physicians in charge are to collect data on adverse reactions by
the following procedures.
Procedure 1:
In interviews with the
subjects, encourage them to report spontaneously on any adverse reactions by
asking questions (e.g., “Have you had any symptoms that bothered you in the
last xx weeks?”).
Procedure 2:
Verify if they have had any symptoms through interviews and using checklists. Limit the symptoms to be verified here to the following four symptoms: hypoglycemia, edema, palpitation, and shortness of breath.
Procedure 3:
Identify symptoms
not perceived by the subjects by auscultation and percussion. Additionally,
identify changes in the laboratory and vital signs data that may be clinically
significant or meaningful. The physician in charge may use his/her judgment in
deciding which change (e.g., in laboratory findings) may be construed as “clinically
meaningful”.
The physician in
charge is to handle symptoms and signs that newly occurred or worsened after
the start of the study as adverse reactions, of all symptoms, signs and changes
in laboratory findings, and to deal with them appropriately, to follow up on
these symptoms and signs until they disappear, resolve or return to their
baseline status, and to describe the name(s) of the adverse reaction(s), date
of their occurrence, their extent, their causal relationship to the
investigational treatment, their severity, and how they were dealt with (e.g.,
the investigational drug was reduced or discontinued or switched to another
drug), their outcome, and date their outcome confirmed in a case report form.
In the event that
the physician in charge judged follow-up unnecessary before these symptoms and
signs disappear, resolve or return to their baseline status, he/she is to give
reasons for such decision in the case report form.
Rationale for the
data collection procedures
These procedures
were introduced, on the grounds that the incidence of adverse reactions could
vary greatly depending on whether it draws on spontaneous reporting by the
subjects (passive surveillance) or on interview- or checklist-based reporting
by the physicians (active surveillance) for data collection. In the current
multi-center trial, safety surveillance data would not be integrated were it
not for the procedures for data collection for use by all participating
institutions. While data on adverse reactions are usually collected by passive
surveillance in studies such as this one, it was decided that active surveillance
be employed in this study to collect data on four major symptoms, namely
hypoglycemia (particularly in elderly patients), edema, palpitation and
shortness of breath to detect signal symptoms of cardiac failure as serious
drug-induced adverse reactions, as the study aims to implement rigorous
glycemic control and involves the use of thiazolidinedione derivatives.
5.3 Severity of adverse reactions and their causal relationship to the investigational treatment
If an adverse
reaction occurs, the physician in charge is to rate its severity by using the
following three-grade criteria.
|
Mild |
Symptoms or
signs are present but the subject experiencing them does not require
treatment to continue with the study |
|
Moderate |
Symptoms or
signs are present but the subject experiencing them calls also for dose
reduction of the investigational drug or addition of another drug to continue
with the study |
|
Severe |
The subject
experiencing the symptoms and signs is having difficulty performing daily
activities, or his/her clinical course is seriously affected by the symptoms
and signs, and the investigational drug needs to be discontinued. |
Then, the
physician in charge is to rate the casual relationship of the adverse reaction
to the investigational drug by using the following four-grade criteria. When
the physician in charge judged it to be “unrelated”, he/she is to describe the
reasons for such judgment.
|
Unrelated |
The adverse
reaction can be clearly accounted for by other factors than the
investigational drug. |
|
Possibly related |
While the
adverse reaction is assumed to have resulted from other factors than the
investigational drug, its causal relationship cannot entirely be ruled out. |
|
Probably related |
The adverse
reaction is least likely to have resulted from other factors than the
investigational drug. |
|
Definitely
related |
The adverse
reaction is highly likely to have resulted from the investigational drug,
e.g., there is a time-dependent association between the time the
investigational drug was given and the occurrence of the adverse reaction, or
the adverse reaction cannot be accounted for by other reasons than the use of
the investigational drug. |
All adverse
reactions except those defined as “unrelated” are to be handled as side
effects.
5.4 Serious adverse reactions
5.4.1 Technical terminology
Any adverse
reaction is defined as serious adverse reactions, if it:
Is fatal
Is life-threatening
Leads to the subject becoming hospitalized or calling for prolonged
hospitalization or treatment
Leads to permanent or serious damage or dysfunction in the subject
Causes congenital anomaly
Is serious hypoglycemia accompanied by impaired consciousness
Is some other serious medical phenomenon
Or is serious enough to place the subject in a critical situation
calling for treatment to avoid the outcomes described above, while it is not
immediately fatal, life-threatening or leads to the subject being hospitalized.
5.4.2 Reporting procedure
When a serious
adverse reaction occurred, irrespective of its causal relationship to the
investigational drug, the physician in charge is to report the adverse reaction
to the Trial Secretariat Office within 72 hours of his/her knowledge of its
occurrence.
In this regard, the deadline
for reporting to the supervisor at his/her own healthcare institution is to be
consistent with the regulations laid down by the institution for such
reporting. Similarly, all participating healthcare institutions are to report
spontaneously to the Pharmaceutical and Food Safety Bureau, Ministry of Health,
Labor and Welfare (MHLW), in accordance with the MHLW undertaking, the “Pharmaceuticals
and Medical Device Safety Information Reporting System”, as well as to relevant
pharmaceutical companies in accordance with the “Pharma Reporting System” based
on the Pharmaceutical Affairs Law.
5.4.3 Assessment by the Research leader and the Trial Secretariat Office
In the even that
the research leader receives case reports on adverse reactions, he is to assess
their urgency, importance or implications, and to give instructions the Data
Center and the participating healthcare institutions of interest, as to their
handling, including putting these cases on hold, as required. The research
leader may communicate these instructions by telephone depending on the urgency
of the cases, followed by those in writing (i.e., by facsimile, mail or
e-mail).
The research leader is to
communicate his interpretation of the adverse reactions reported as well as
their handling (including judgment as to continuation or termination of the
study) to the research leader within 72 hours of their knowledge of occurrence
of the adverse reactions, to ask for judgment regarding the validity of their
judgment and handling of the cases and forward the “Case Report on Adverse
Reactions” sent to them by the reporting healthcare institutions. Assessment of
serious adverse reactions by the research leader should include an evaluation
of the each subject’s clinical course, as well as of whether or not the
frequency of each reported adverse reaction is within their anticipated range. If
the research leader judged their frequency to be beyond their anticipated range,
the research leader is to document such judgment for reporting.
The research leader is to
submit all reported adverse reactions to the Safety Assessment Committee for
review. The Safety Assessment Committee is to review all reported adverse
reactions, and to make recommendations to the research leader with regard to
the handling of the cases (including judgment as to whether the study is to be
continued with the subjects, instructions are required to the healthcare
institutions of interest, or the trial protocol calls for revision). The
research leader is then to give instructions to the research leader in
accordance with these recommendations.
5.5 Analysis of adverse reactions through regular monitoring
The research
leader and the Trial Secretariat Office are to analyze monitoring reports
produced by the Data Center through regular monitoring, and to develop reports
on the results of their analysis, where all adverse reactions not reported as
urgent are to be evaluated with regard to their category, severity, and rate of
occurrence, to determine if they require reporting to the healthcare
institutions of interest or call for revision of the trial protocol. At the
same time, the research leader and the Data Center are to ensure: that no adverse
reactions are left out/missed in the reports submitted by the healthcare
institutions; that all reported adverse reactions are entered in regular
monitoring reports; and that the results of their analysis include their
judgment as to whether there were any adverse reactions left out/missed in the
reports received from the participating healthcare institutions.
5.6 Adverse reactions to be anticipated
All investigators
are to refer to the package inserts and the product monographs of the drugs to
be used in the trial for information on anticipated adverse reactions with the
drugs as well as their rate of occurrence.
Specifically,
anticipated adverse reactions include hypoglycemia, edema, cardiac failure,
gastrointestinal symptoms including nausea/vomiting and discomfort, anemia,
palpitation, skin rash, eczema, high CPK values, rhabdomyolysis, lactic
acidosis, leucopenia, thrombocytopenia, hyperkalemia, and abnormal GOT/GPT
values.
6. Target patient accrual and the rationale for the target setting
The target patient
accrual is projected at 1,669 each for intensive and conventional therapy,
totaling 3,338 patients involving a total of 81 healthcare institutions that
each allows 50 or more subjects to be accrued.
The
target patient accrual number was calculated on the following rationale.
The
trial subjects were estimated to include those with ischemic heart disease and
those without at a ratio of 7:3, with the occurrence of events including deaths
per year estimated as 4.4% (10.0% in those with a history of ischemic heart
disease and 2.0% in those without) in the conventional therapy arm versus 3.08%
(7.063% in those with a history of ischemic heart disease and 1.395% in those
without) in the intensive therapy arm.
Based
on this estimate, under the assumption that the subjects are to be accrued over
a period of 1 year for follow-up for 2 years and 9 months, the number of
patients required for this trial was calculated by using the Shoenfeld-Richter
nomograms as 1,408 subjects per arm with the expected number of events being
328, to verify that intensive therapy is superior to conventional therapy at
the two-sided significance level of 5% with a 90% power. In light of the
results of the general survey conducted in June 2007, the annual event rate was
revised to about 1/2 of the original estimate, with the hazard ratio estimated,
as previously estimated, as 2.2% in the conventional therapy arm versus 1.5347%
in the intensive therapy arm. With the patient accrual extended to December
2008 (with an accrual period of 2.5 years) and follow-up extended to March 2013
(with a follow-up period of 4.25 years) based on this estimate, the expected
number of events remained the same, and the number of subjects required for
both arms were estimated to total 2,338.
However,
in the event that the expected number of events is to be judged unattainable
given the pace at which events occurred in both arms at the time of regular
monitoring by the Data Center, the Trial Secretariat is to consult the trial
statisticians and file a request for revision of the trial protocol with the
Central Ethics Committee, where revision of the trial protocol is to be
construed as either of the following:
Extension of the patient accrual period or the follow-up period
‚ Revision upward of the patient accrual number
The
final number of subjects who enrolled in the study during the registration
period was 2,542 patients. Based on a patient accrual period of 2.5 years with
a follow-up period of 4.25 years, the power of the test will be ³
80% with a 5% level of significance (2-sided). Therefore, the expected number
of events will be changed to 250 because an interim analysis aimed at
demonstrating the superiority of intensive therapy over conventional therapy in
order to discontinue the trial, has not been conducted.
The
rate of occurrence was lower than what was expected before the start of the
study, suggesting the number of events would not reach the required number of
events during the predefined follow-up period. Since the registration period
was expired, addition of subjects was impossible. Thus, follow-up period was
extended for a sufficient time until the required number of events could be
obtained.
7. Committees
7.1 Trial Coordinating Committee
The research leader may delegate to the Trial Coordinating Committee
authority in coordinating the interpretation of the trial protocol and other
minor details of the study.
The coordination the research leader may delegate to the Trial
Coordinating Committee includes the following:
1) Coordination among the participating healthcare institutions about the
minor details of the trial protocol
2) Harmonization of potential conflicting interpretation of the trial
protocol during conduct of the trial
3) Coordination among the participating healthcare institutions
The Trial Coordinating Committee is to coordinate among the healthcare institutions
about the minor details of the trial protocol based on opinion and feedback
gathered from these institutions. Additionally, when the trial protocol calls
for revision, the Trial Coordinating Committee is to receive feedback on the
proposed revision of the trial protocol from the participating institutions and
to make the revised protocol definitive in light of this feedback.
7.2 Trial Administrative Committee
The Trial Administrative Committee is to be composed of 15 committee
members to be appointed by Director of the Japan Diabetes Foudation (JDF), who
provide consultation and make recommendations regarding the following in
response to requests by Director of the JDF:
(1) Evaluation of researcher participant applicants for the strategic
outcome research
(2) Alignment of the trial conduct infrastructure
(3) Planning of the strategic outcome research plan
(4) Budgeting and audit of the strategic outcome research project
(5) Other fundamental issues in the implementation of the strategic
research project
The Chairperson of the Trial Administrative Committee is to be appointed
by Director of the JDF.
The Director of the JDF is to report to the Trial Administrative
Committee on the results of deliberations and notifications by all the trial
committees (except for the Trial Administrative Committee).
7.3 Trial Assessment Committee
The Trial Assessment Committee is to provide interim reports (evaluations
of interim analyses) on the progression of the trial in view of the
recommendations it is to make regarding the continuation of the trial or
release of the study results while being in close contact with the Trial
Progress Management, and to make recommendations regarding the measures and
actions to be taken to the research leader and the JDF Secretariat.
The
Chairperson of the Committee is to be chosen by the members of the Committee
from among the members of the Committee. The Chairperson of the Committee is to
convene members of the Committee and to serve as Chairperson. Meetings by the
Committee are to be held with the JDF Secretariat, Trial Progress Management
members, the research leader and research leader-appointed personnel (including
the independent statistical analyst) attending, in addition to the members of
the Trial Assessment Committee.
The
Chairperson is to appoint a committee member in charge of the meeting minutes
prior to the meeting, where the Chairperson may combine this role. Prior to
deliberations of a particular research issue, the research leader is to provide
an outline of the issue, along with any new developments with the issue.
Members of the Trial Progress Management are to report on the status of trial
monitoring. Prior to deliberations, all members of the Trial Assessment
Committee and the independent statistical analyst are to excuse themselves, and
the independent statistical analyst is to account for the interim analysis
results and evaluate the results. The Trial Assessment Committee is to make its
recommendations as to the “early termination” or “continuation” of the study
after its consensual decision making. If the decision was divided, the decision
is to be made by a majority vote, where in the case of a tie the Chairperson is
to cast a tie-breaking vote. The committee member in charge of the meeting
minutes is to summarize the results of deliberations, and to seal these minutes
together with the interim analysis results. After deliberations, the Committee
is to notify the research leader and the JDF Secretariat of their
recommendations. The interim analysis results and the minutes of the Committee
deliberations thus sealed are not to be unsealed for disclosure until trial termination
is recommended or until the completion of the final analysis.
7.4 Trial
Progress Management Committee
The Trial Progress Management is to audit monitoring of the clinical
trial except in the interim analyses. This monitoring refers to central
monitoring as conducted via the
The
Chairperson of the Committee is to be appointed by Director of the JDF.
The
Trial Progress Management may be run by means of consensual decision making
through teleconferencing, communications by post, e-mails, in addition to
on-site meetings.
The
Trial Progress Management is to receive as month updates from the Data Center
on the patient accrual status, patient eligibility for study entry,
appropriateness of subject allocation, the presence or absence of trial
violations, the incidence of serious adverse reactions, and the occurrence of
endpoints with the treatment arms masked. The Trial Progress Management is to
evaluate whether the study is being performed in a safe and appropriate
fashion, and to report to the research leader on the results of their
deliberations.
If
there occurred a problem with regard to the progress of the study that might
affect the science and ethics of the study, the Trial Progress Management is to
report to the research leader to that effect, and to discuss solutions to the
problem with the relevant research groups and research support organizations.
7.5 Safety Assessment Committee
In response to requests from the research leader, the Safety Assessment
Committee is to discuss the measures and actions to be taken as well as the
casual relationship with the investigational treatment in regard to the adverse
reactions for which emergency reporting is designated, and to report to the
research leader.
The
Chairperson of the Committee is to be appointed by Director of the JDF.
When
an adverse reaction is reported that require evaluation by the Safety
Assessment Committee, the research leader is to make an immediate request to
the Committee Secretariat to review the adverse reaction, and the Committee
Secretariat is to communicate the reported status to 2 or more members of the
Committee by post or by parcel delivery service.
When
the research leader and the Safety Assessment Committee Secretariat found the
reported details inadequate, they may ask the reporting physician for a greater
account of the adverse reaction reported.
Upon
request from the research leader, the Safety Assessment Committee is to provide
the results of their deliberations on the case submitted, and the Chairperson
is to summarize these deliberations. When the Chairperson found the deliberations
to possibly affect the decision as to the continuation of the study or call for
revision of the trial protocol, he/she is to report to the research leader to
that effect immediately. When the above considerations do not apply, the
Chairperson is to report to the research leader in his monthly reports.
When
the research leader received a report from the Safety Assessment Committee on
an adverse reaction reported whose causal relationship with the investigational
treatment could not be denied, the research leader is to communicate the review
to the Trial Secretariat immediately, and give instructions on the measures and
actions to be taken.
7.6 Central Ethics Committee
JDF is to set up the Central Ethics Committee to ensure the conduct of
the medical research in accordance with the Helsinki declarations.
The Central Ethics Committee is to be comprised of the following
personnel to be appointed by Director of the JDF.
l
1 Chairperson
l
Members of the Committee
including those that are given as follows:
1) 2 or more experts in clinical trials
2) 2 or more medical experts
3) Non-medical experts
1
or more experts in humanities and social sciences such as experts in legal
science
1
or more representatives of the general public
4) Other personnel to be appointed by the Director of the JDF.
The Central Ethics Committee is to review the ethical and scientific
validity of any medical research that comes within its purview at the request
of the Director of the JDF, and to make relevant recommendations in writing.
Additionally, the Central Ethics Committee is to review reports by the Trial
Assessment Committee, the Safety Assessment Committee, the Trial Progress
Management and the intramural Ethics Review Committee of the participating
healthcare institutions as to their ethical and scientific validity, and to
make recommendations regarding the continuation, revision, or early termination
of the study.
